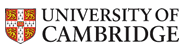

Visualizing basins of attraction for different minimization algorithms
The assignment of an atomic configuration to a basin of attraction depends on how the potential energy is minimized. A survey of several minimization algorithms shows that basins may be compact or disconnected with complex boundaries.

Self-assembled fibrils of patchy spherocylinders
Proteins can aggregate in a wide variety of structures, both compact and extended. Simulations of a coarse-grained anisotropic model have reproduced many of the experimentally observed aggregate structures.
Design rules for DNA-coated nanoparticles
Monte Carlo simulations predict the phase behavior of colloids coated with long, flexible DNA chains. An important change occurs in the phase diagram when the number of DNAs per colloid is decreased below a critical value.

Designing super-selective nanoparticles
Simulations of “multivalent” nano-particles that can bind to a large number of ligands simultaneously display regimes of “super selectivity” where the fraction of bound particles varies sharply with the receptor concentration.
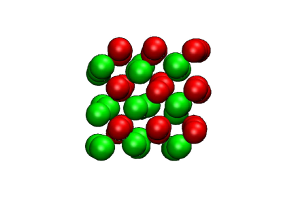
Direct determination of the size of basins of attraction of jammed solids
A free-energy-based Monte Carlo method can be used to estimate the number of distinct potential-energy minima in jammed solids, even when this number is much too large to be sampled directly.
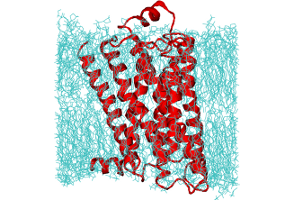
Ligand binding cooperativity of membrane receptors
Signal transduction upon binding of a ligand to a membrane protein can occur through fluctuations; a numerical study demonstrates the influence of conformational fluctuations on the cooperativity of a binding reaction.

Simulation of nucleation in almost hard-sphere colloids
Crystal nucleation rates calculated for the Weeks-Chandler-Andersen potential agree well with the nucleation rates predicted for hard spheres although at low supersaturation they disagree strongly with experimental results.
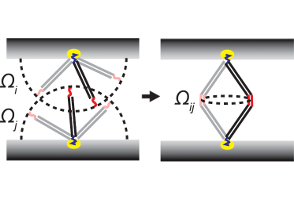
Pair potentials for DNA-coated micro- and nanoparticles
DNA-coated colloids have great potential for the design of complex self-assembling materials. In order to predict the structures that will form, knowledge of the interactions between DNA-functionalized particles is crucial.
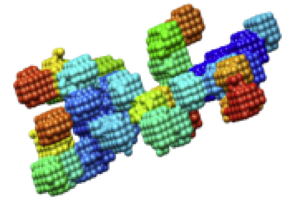
Protein-solvent interactions and protein aggregation
The folding specificity of proteins can be simulated using simplified structural models and knowledge-based pair potentials. When the same models are used to simulate systems containing many proteins, large aggregates tend to form.
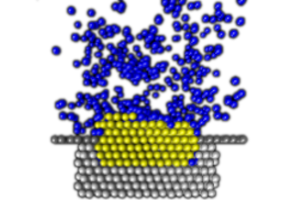
Nanoscale pits for nucleating protein crystals
Growing high-quality crystals is a bottleneck in determining protein structures by x-ray diffraction. Computer simulations report rapid crystal nucleation in nanoscale pits, as found in experiments with disordered pitted surfaces.
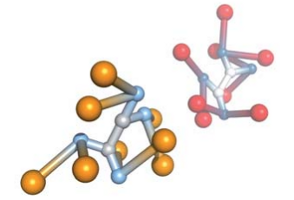
Phase transitions in binary mixtures of DNA-coated particles
With strong hybridization, a binary mixtures of DNA-coated particles with “sticky” ends undergo a phase separation between a dilute vapor-like phase and a dense network-forming liquid-like phase.
Modelling and simulation of electrokinetic effects
Electrokinetic phenomena are of great practical importance in fields as diverse as micro-fluidics, colloid science and oil exploration. However, the quantitative prediction of electrokinetic effects has been limited to relatively simple geometries.